Predicting and designing proteins (1): How protein structure became a computational problem
Recently I’ve been working on predictive and generative models of proteins. Quite a shift in expertise, if you ask me! Therefore, I may as well write some literature review posts about proteins here. (Not that I have no more SWE knowledge to share, and not that AI has fully replaced us for non-cutting-edge material to write about. Well... 🙁)
P.S. I’m sorry for not updating anything since March; consider this series me making good on my earlier promise to update more often.
A protein is a chain whose units are called amino-acid residues. “Residue” is the conventional name for an amino acid after it has been joined to the chain. Each residue has the same backbone pattern and one of twenty common side chains, which differ in size, charge, flexibility, and chemical reactivity. The order of those residues is the protein’s sequence. The chain can bend around its backbone bonds, bring distant residues together, exclude water from some regions, expose other regions to solvent, and form a three-dimensional arrangement that biologists call a structure or fold.
The backbone can be pictured as the repeating cord of the chain, while the side chains are chemically different attachments projecting from it. Water-avoiding side chains tend to become buried together, charged side chains often remain exposed to water or pair with opposite charges, and hydrogen bonds help stabilize recurring backbone arrangements. None of these preferences acts as a rigid instruction. The final fold emerges from many individually weak interactions being satisfied, frustrated, or traded against one another at the same time.
The central computational problem is easy to state: given the sequence, what structures will the chain adopt? The simplicity of the question is misleading. A protein is not folded by placing one residue at a time into a rigid final sculpture. Its atoms move in a warm solvent, many weak interactions act at once, and the chain can visit an enormous number of arrangements. A useful prediction must therefore identify a narrow set of biologically relevant structures without explicitly testing every geometrical possibility.
What Anfinsen’s experiments established
In the middle of the 20th century, it was not obvious where the instructions for folding resided. A cell contains molecular machinery that synthesizes proteins and other molecules that assist folding. One possibility was that a protein required an external template to acquire its shape. Experiments on the enzyme ribonuclease A helped narrow the alternatives. Christian Anfinsen and colleagues unfolded the enzyme and disrupted its disulfide bonds, then showed that suitable conditions allowed it to recover both its structure and catalytic activity. Anfinsen’s later synthesis expressed the thermodynamic hypothesis: under an appropriate environment, the native conformation is determined by the amino-acid sequence and corresponds to a favourable free-energy state.1
A disulfide bond is a covalent link between two cysteine side chains; breaking several such links removes structural fasteners as well as the weaker interactions disturbed during unfolding. An enzyme is a molecule that accelerates a chemical reaction, so the return of ribonuclease activity was an especially informative test: the recovered chain had not merely become compact again, but had restored the precise arrangement needed to perform chemistry. “Favourable free energy” means that, among the accessible alternatives under those conditions, the folded population is thermodynamically preferred. It does not mean that every individual protein molecule is motionless or that only one conformation ever exists.
This was an enabling claim, not a complete theory of folding. It made sequence-to-structure prediction meaningful because the required information was, in principle, present in the sequence. It did not say that a protein has only one biologically relevant shape, that cells never use folding helpers, or that the chain finds its native state by exhaustively trying every possibility. It also did not provide an efficient algorithm. The number of possible backbone arrangements grows so rapidly that a naive search would remain impossible even with very fast computers.
The distinction between a thermodynamic statement and an algorithmic one shaped the next several decades. One research programme tried to approximate physical forces and search the resulting energy landscape. Another tried to reuse structures already discovered by evolution and measured by experiments. Modern learned models draw from both programmes.
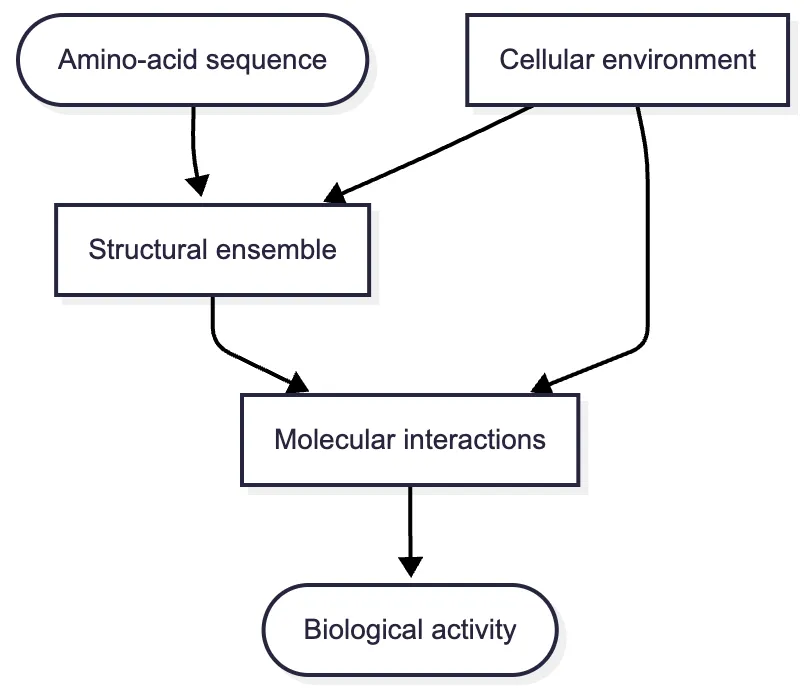
The diagram includes an ensemble rather than one structure. An ensemble is a collection of conformations with different probabilities. Many early computational methods sought a single representative structure because that was already difficult and because experimental databases were dominated by deposited coordinate sets. The later importance of dynamics does not make those static models useless; it clarifies what they approximate.
Why a structural archive changed the problem
Prediction requires examples against which ideas can be tested. X-ray crystallography, nuclear magnetic resonance spectroscopy, and later cryogenic electron microscopy made it possible to infer atomic coordinates experimentally. The Protein Data Bank (PDB), founded in 1971, turned those results into a shared, machine-readable archive.2 Its importance was not simply that it stored pictures. Each entry connected a biological molecule, an experimental procedure, a sequence, and a set of coordinates. That made structural biology cumulative and eventually made supervised learning possible.
These techniques observe structure indirectly. Crystallography infers an electron-density pattern from how a crystal scatters X-rays; nuclear magnetic resonance uses signals from atomic nuclei to constrain distances and motions in solution; cryogenic electron microscopy reconstructs three-dimensional density from many images of rapidly frozen particles. Researchers then build atomic models into those measurements. A PDB coordinate is therefore an experimentally supported model with a resolution and uncertainty, not a literal photograph of stationary atoms.
The archive revealed a useful regularity: evolution often changes sequence more quickly than it changes the overall fold. Two proteins can descend from a common ancestor and retain similar structures despite many residue substitutions. Such proteins are homologues. If a query sequence resembles a protein with a known structure, the known structure can serve as a template. Chothia and Lesk quantified the relationship between sequence divergence and structural divergence, giving comparative modelling an empirical basis.3
Comparative, or homology, modelling then became a structured inference problem. One first identifies a template, aligns the query residues to the template residues, transfers corresponding geometrical restraints, builds unaligned loops and side chains, and refines the result. MODELLER formalized much of this process as the satisfaction of spatial restraints.4 Threading methods widened the search by asking whether a sequence was compatible with the residue environments of a known fold even when obvious sequence similarity had disappeared.5
These methods were powerful precisely because they did not solve folding from first principles. They exploited the fact that evolution repeatedly modifies existing proteins and that the archive already contained many reusable folds. Their limitation followed from the same source: when no suitable template existed, the search became much harder.
How physical search became practical enough to design with
Template-free methods approached the chain as a physical and statistical object. The Rosetta project, developed in David Baker’s group, began from an observation about local structure: short sequence fragments often adopt similar local conformations in known proteins. Rosetta assembled such fragments into candidate backbones and used a scoring function to favour globally coherent arrangements.6 Later versions used more detailed all-atom energy terms to represent packing, hydrogen bonding, electrostatics, and solvation.7
The key idea was not that fragment assembly reproduced the literal path by which a protein folds. It was a search strategy. Local fragments restricted the possibilities; the energy function ranked completed structures; repeated stochastic trials explored different combinations. The method exchanged an impossible exhaustive search for a guided but still expensive one.
Rosetta also made the reverse question approachable. If a backbone is specified, which sequence would make that structure energetically favourable? In 2003, Brian Kuhlman and colleagues reported Top7, a designed globular protein with a topology not previously observed in nature. Its experimental structure closely matched the computational model.8 The result mattered because it connected calculation to synthesis and structural measurement. The software had not merely recognized a naturally evolved fold; it had helped specify a new one.
Subsequent work distilled recurring geometric rules for “ideal” proteins, including preferred loop lengths and ways of packing helices and sheets.9 These rules reduced the search space and made design more systematic. They also exposed a theme that returns throughout this series: good generative methods do not sample every mathematically possible object. They encode constraints that make useful objects much more likely.
What the early programs actually computed
The names “comparative modelling” and “physical search” can make these systems sound less concrete than they were. MODELLER began with an alignment between a query sequence and one or more template structures. From corresponding template residues it constructed probability distributions for distances, bond angles, and dihedral angles in the query. These became spatial restraints: numerical penalties that grew when a candidate model violated the geometries expected from the templates. An optimizer then changed the query coordinates to reduce the sum of restraint violations and stereochemical penalties.4 The program did not copy a template atom for atom. It translated uncertain correspondence into an objective function and searched for a structure that jointly satisfied many imperfect constraints.
Rosetta used a different representation of uncertainty. For each short window of the query—originally fragments of three and nine residues—it searched the structural database for fragments with compatible local sequences and secondary-structure predictions. A trial move replaced the current backbone angles of one window with those from a library fragment. Monte Carlo sampling accepted moves that improved the score and sometimes accepted worse moves, especially early in simulated annealing, so the search could escape a poor local arrangement. Thousands of independent trajectories were needed because any one path could settle in the wrong basin.67
Early Rosetta also separated two resolutions. A coarse “centroid” stage represented each side chain approximately, which made large backbone moves inexpensive. Promising backbones then entered an all-atom stage, where side chains were assigned discrete conformations called rotamers and repacked while a more detailed energy function scored steric clashes, hydrogen bonds, electrostatics, solvation, and torsional preferences. This division addressed a practical conflict that persists in generative modelling: broad exploration is easier with a simplified representation, while final discrimination requires chemical detail. It did not remove the dependence on the scoring function. If an incorrect compact structure received an artificially favourable score, more sampling could make the wrong answer easier to find rather than correct it.
How neural networks entered before end-to-end folding
Machine learning was part of protein prediction decades before AlphaFold. The early target was usually secondary structure: local backbone patterns such as alpha helices, beta strands, and irregular coil regions. Predicting these categories is easier than placing every atom, but it tests whether a model can infer structural tendencies from sequence context.
In 1988, Ning Qian and Terrence Sejnowski trained neural networks to predict secondary structure from local sequence windows.10 The networks were small by modern standards and had no direct representation of global three-dimensional geometry. Their importance lay in demonstrating that nonlinear learners could recognize recurring sequence–structure relationships from examples.
PSIPRED later made a large practical improvement by supplying a neural network with position-specific profiles derived from related sequences.11 Each profile summarized which amino acids appeared at one aligned position and how strongly they were conserved. The combination anticipated the next era: evolutionary preprocessing produced an informative representation, and a learned model converted it into structural predictions.
At the implementation level, PSI-BLAST first searched a sequence database for relatives and assembled the profile as a position-specific scoring matrix. One axis indexed positions in the query; the other recorded how compatible each amino-acid type was with each position. A feed-forward neural network read overlapping windows of this matrix and produced three scores per central residue—for helix, strand, or coil. A second network examined the first network’s outputs across neighbouring positions and corrected locally implausible patterns, such as an isolated one-residue helix.11 This two-stage system could learn local regularities, but its windowed inputs and three output labels could not represent the full set of long-range constraints needed to place a protein in three dimensions.
How blind evaluation changed scientific progress
A prediction method can appear successful if it is tested on structures that influenced its design, if easy examples dominate the benchmark, or if failures remain unpublished. The Critical Assessment of protein Structure Prediction (CASP), initiated by John Moult and colleagues in 1994, addressed these problems with a recurring blind experiment. Organizers obtained sequences whose experimental structures were not yet public; modelling groups submitted predictions; and independent assessors compared predictions with the subsequently released structures.12
CASP gave the community more than a leaderboard. It separated template-rich from template-poor targets, made improvements visible under common conditions, and forced researchers to confront generalization. The competition also generated a history of changing bottlenecks. Better template detection and fragment assembly improved some categories; evolutionary information later transformed difficult targets; deep learning then altered the entire prediction pipeline.
Blind evaluation remains essential even though the data environment has changed. Today, a model may have seen billions of sequences, hundreds of millions of predicted structures, and many versions of the PDB. Excluding an identical test structure is no longer sufficient if close homologues, the same structural motif, or predictions derived from that target remain in training data. CASP’s durable contribution is the principle that evidence should be separated from the process that produced the model.
Conclusion
By the end of the 20th century, the field possessed the conceptual ingredients needed for machine learning: a sequence-to-structure premise, a growing archive of labelled examples, physical scoring functions, reusable structural templates, early neural networks for local secondary structure, and blind evaluation. What it still lacked was a way to infer long-range relationships reliably when no close template existed.
The next advance came from a source that had been present all along: evolution. A sequence database does not only contain many individual proteins. It contains the record of many related proteins changing together. Learning how to read that record turned sequence variation into approximate geometric information.
Onto the next article.
References
Anfinsen, C. B. (1973). Principles that govern the folding of protein chains. Science, 181(4096), 223–230. https://doi.org/10.1126/science.181.4096.223↩
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., et al. (2000). The Protein Data Bank. Nucleic Acids Research, 28(1), 235–242. https://doi.org/10.1093/nar/28.1.235↩
Chothia, C., & Lesk, A. M. (1986). The relation between the divergence of sequence and structure in proteins. The EMBO Journal, 5(4), 823–826. https://doi.org/10.1002/j.1460-2075.1986.tb04288.x↩
Šali, A., & Blundell, T. L. (1993). Comparative protein modelling by satisfaction of spatial restraints. Journal of Molecular Biology, 234(3), 779–815. https://doi.org/10.1006/jmbi.1993.1626↩
Bowie, J. U., Lüthy, R., & Eisenberg, D. (1991). A method to identify protein sequences that fold into a known three-dimensional structure. Science, 253(5016), 164–170. https://doi.org/10.1126/science.1853201↩
Simons, K. T., Kooperberg, C., Huang, E., & Baker, D. (1997). Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. Journal of Molecular Biology, 268(1), 209–225. https://doi.org/10.1006/jmbi.1997.0959↩
Rohl, C. A., Strauss, C. E. M., Misura, K. M. S., & Baker, D. (2004). Protein structure prediction using Rosetta. Methods in Enzymology, 383, 66–93. https://doi.org/10.1016/S0076-6879%2804%2983004-0↩
Kuhlman, B., Dantas, G., Ireton, G. C., Varani, G., Stoddard, B. L., & Baker, D. (2003). Design of a novel globular protein fold with atomic-level accuracy. Science, 302(5649), 1364–1368. https://doi.org/10.1126/science.1089427↩
Koga, N., Tatsumi-Koga, R., Liu, G., Xiao, R., Acton, T. B., Montelione, G. T., & Baker, D. (2012). Principles for designing ideal protein structures. Nature, 491(7423), 222–227. https://doi.org/10.1038/nature11600↩
Qian, N., & Sejnowski, T. J. (1988). Predicting the secondary structure of globular proteins using neural network models. Journal of Molecular Biology, 202(4), 865–884. https://doi.org/10.1016/0022-2836%2888%2990564-5↩
Jones, D. T. (1999). Protein secondary structure prediction based on position-specific scoring matrices. Journal of Molecular Biology, 292(2), 195–202. https://doi.org/10.1006/jmbi.1999.3091↩
Moult, J., Pedersen, J. T., Judson, R., & Fidelis, K. (1995). A large-scale experiment to assess protein structure prediction methods. Proteins: Structure, Function, and Genetics, 23(3), ii–iv. https://doi.org/10.1002/prot.340230303↩